

EGFR突变型肺癌靶向治疗的耐药机制及其对策,目前肺癌的发病率位居恶性肿瘤首位,其中非小细胞肺癌(non-small cell lung cancer,NSCLC)占肺癌总发生率的85%,提高此类疾病的治疗反应率、延长患者的生存时间和建立新的治疗方式十分重要。
EGFR突变型肺癌靶向治疗的耐药机制及其对策
目前肺癌的发病率位居恶性肿瘤首位,其中非小细胞肺癌(non-small cell lung cancer,NSCLC)占肺癌总发生率的85%,提高此类疾病的治疗反应率、延长患者的生存时间和建立新的治疗方式十分重要。
近年来靶向治疗成为NSCLC的治疗的重要组成部分,其中表皮生长因子受体(epidermal growth factor receptor,EGFR)是目前备受瞩目的肿瘤治疗靶点,针对其靶向治疗越来越受到人们的重视,靶向治疗正在改变晚期NSCLC的治疗模式。
同时也引发一些新问题,吉非替尼、厄洛替尼等EGFR抑制剂(EGFR-TKI)针对EGFR突变晚期NSCLC取得了令人瞩目的疗效,以及随后发现EGFR-TKI在治疗NSCLC时的原发性耐药或继发性耐药,是我们在治疗晚期NSCLC面临新的挑战,继而开展新的探索,寻找对策。
1.EGFR的结构与功能
EGFR由高度保守的胞内酪氨酸激酶区、胞外配体结合区和单链跨膜区组成,当配体结合区与配体结合后,表皮生长因子受体可由单体转化为二聚体,使得体内的酪氨酸激酶自身磷酸化,进一步激活下游的信号通路,从而最终导致细胞的增殖和生存(如下图)。
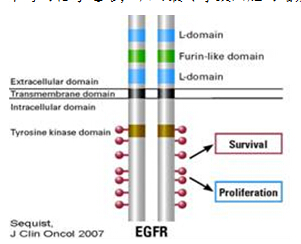
图1: EGFR是一种跨膜受体,EGF结合于胞外结构域即形成受体二聚体并激活细胞内酪氨酸激酶结构域,引发激酶自磷酸化和下游分子的磷酸化,激活包括增殖和存活在内的多种细胞功能。
2.EGFR突变及EGFR酪氨酸激酶抑制剂的作用机制
目前现已报道EGFR突变主要有如下几类: (1)19位外显子突变主要是第746-752位密码子的缺失突变,导致EGFR蛋白中氨基酸序列丢失,改变了受体ATP结合囊(ATP-binding poke,ABP)角度,显著增强了靶向药物对肿瘤细胞的敏感性,该类突变占55%。
(2)21外显子的突变均为错义突变,突变主要发生在858位的L即精氨酸突变为R即亮氨酸(L858R),此突变位点紧邻激酶活化环(activation loop)中高度保守的模体,增强了肿瘤细胞对EGFR—TKI的敏感性,该类突变占40%。⑶其他位点的突变约占5%(图2)。上述EGFR 突变多靠近ATP 结合位点,因而影响受体的激酶活性,使细胞处于持续的活性状态,导致细胞异常增殖及抗凋亡。
EGFR酪氨酸激酶抑制剂(EGFR-TKI)是针对EGFR的分子靶向药物,主要通过与ATP竞争性结合位于细胞表面的EGFR酪氨酸激酶催化域结合位点,阻断信号向细胞内的进一步传递,抑制肿瘤细胞生长并诱导其凋亡。目前厄洛替尼、吉非替尼等EGFR-TKI已广泛用于临床。
分子生物学研究已证实具有EGFR 突变的肺癌细胞对EGF-TKI的治疗敏感,且19位外显子变异较L858突变对EGFR-TKI更加敏感,上述敏感性的增加一方面可能是由于EGFR 突变细胞对EGFR 信号通路的依赖性较前增强,该通路被阻断时更强烈影响细胞的生长;另一方面的原因可能是因为突变改变了EGFR 内关键的氨基酸残基的结构,使EGFR与EGFR-TKI更紧密结合,对酪氨酸激酶活性的抑制作用增强。
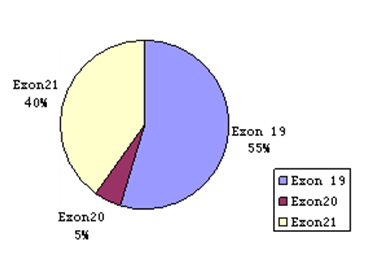
图2:东亚裔人群中EGFR突变类型及其所占的百分比(Zhou Q,J Clin Oncol, 2011)。
3.EGFR-TKI的耐药机制
3.1 原发性耐药
大约30%EGFR 突变的NSCLC患者在用EGFR-TKI治疗初期即出现耐药,称之为原发性耐药,原发性耐药的机制主要有两个方面:① K-ras突变导致的原发性耐药;②蛋白酪氨酸磷酸酶基因(PTEN)的缺失导致耐药性的产生。
3.1.1 K-ras突变:K-ras是EGFR下游的一个信号传导通路,两者都与肺癌的发生和治疗密切相关。相对于EGFR的高突变,K-ras的低突变是东亚人群肺腺癌的特征,而西方国家肺癌患者的突变情况则不同,K-ras突变率较高,EGFR突变率较低。
另一点与EGFR突变的不同之处为,K-ras突变与吸烟有明显的相关性,即K-ras突变患者一般都有吸烟史。更重要的是,EGFR突变和K-ras突变不会出现在同一个病例,K-ras基因和EGFR基因是两个独立的致癌因素,有不同的致病机理,但大量的研究表明存在K-ras突变的患者对吉非替尼不敏感,即K-ras突变的患者对EGFR-TKI产生原发性耐药。
3.1.2 PTEN的缺失:PTEN基因编码的蛋白质具有脂质磷酸酶和蛋白磷酸酶活性,发挥双重肿瘤抑制功能。PTEN通过抑制P13K/Akt通路的激活,抑制mTOR的活化。PTEN处于多条信号转导通路的共同节点,若PTEN基因在肿瘤细胞中发生突变、缺失、低表达,就会导致其抑癌功能减弱或丧失。
大量的研究表明PTEN的缺失降低肺癌患者对EGFR-TKI的疗效,功能性的PTEN失活在一些人类肿瘤中普遍存在,如缺乏PTEN蛋白的人NSCLC细胞株H157对吉非替尼耐药,而人NSCLC细胞株H1355(野生型PTEN)对吉非替尼敏感。
下调P13K-Akt活性可使治疗敏感性恢复或提高,mTOR抑制剂雷帕霉素(rapamycin)能够增强PTEN表达缺失的肿瘤细胞对EGFR-TKI的敏感性;将野生型PTEN转染入PTEN表达缺失的肿瘤细胞中,可观察到P13K/Akt活性降低以及对EGFR-TKI的反应性提高,因此PTEN的缺失也是EGFR-TKI原发性耐药的重要因素。
3.1.3 其他原因导致的EGFR-TKI的原发性耐药: 如 PI3K/AKT信号通路的激活; 胰岛素样生长因子1受体(insulin like growth factor 1 receptor, IGF1)介导的信号通路激活;NF-κB信号途径的激活;
EML4-ALK融合基因突变;BRAF基因突变;人表皮生长因子受体-2(human epidermal growth factor receptor-2, HER-2)突变;成纤维细胞生长因子(fibroblast growth factor, FGF)-成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)信号途径;
3.2 继发性耐药
3.2.1 T790M突变:该学说认为EGFR基因第20外显子在应用EGFR-TKI治疗过程中发生了二次突变,导致EGFR 790位上的苏氨酸被甲硫氨酸所取代(T790M)。
一旦苏氨酸被甲硫氨酸取代,其结果在该位点上引入了一条更大的氨基酸侧链构成空间位阻,该空间位阻的形成影响酪氨酸激酶与EGFR-TKI之间氢键的形成,最终导致EGFR-TKI无法与酪氨酸激酶相结合。
Pao等研究者发现在未应用EGFR-TKI治疗的NSCLC患者中,T790M突变的发生率小于0.1%,而对EGFR-TKI继发耐药者中约50%出现T790M突变,该研究成果从另一个侧面反映了EGFR基因的二次突变在EGFR-TKI继发耐药中的地位和作用,即T790M的突变是EGFR-TKI继发性耐药最主要的因素。
3.2.2 MET基因扩增:MET是HGF的受体,编码HGF酪氨酸激酶受体的跨膜区,与肿瘤的侵袭、转移和扩增有关。
Engelman等于2007年首次提出原癌基因MET的扩増是EGFR-TKI的一种继发性耐药机制,他们在构造EGFR-TKI继发性耐药细胞株模型时发现该现象是由MET基因扩增引起的;他们也同时发现有22%的EGFR-TKI继发性耐药患者的肿瘤组织中存在MET基因扩增。
后续的研究表明MET扩增通过激活ERBB3-PI3K信号途径来持续激活下游的信号通路,导致NSCLC对EGFR-TKI产生耐药。不仅如此,Guix等研究也发现扩增MET基因过表达的克隆而发挥对EGFR-TKI的继发性耐药作用,因此,MET基因的扩增在EGFR-TKI的继发性耐药中也具有重要的地位。
3.2.3 HGF过表达:HGF为MET的配体,HGF过表达与EGFR-TKI耐药性相关。给19外显子框内缺失的人肺腺癌细胞予HGF后,细胞对吉非替尼治疗表现出耐药性,且HGF剂量越高,耐药性越强。
3.2.4 HER3磷酸化: erb基因是编码人HER的癌基因,erb1、erb2、erb3和erb4分别编码HER1、HER2、HER3和HER4。HER与配体结合后通过自磷酸化参与细胞的信号传递,最终调节细胞的生长与分裂。
其中HER3本身并无激酶活性,需要与其他的HER家族成员形成异二聚体方能介导信号传递。
Engelman等研究者发现HER3磷酸化使磷脂酰肌醇3激酶(P13K)/Akt通路持续活化而导致肿瘤细胞对EGFR-TKI产生继发耐药。除上述获得性耐药机制之外,还有一些EGFR的二次突变也与癌细胞对EGFR-TKI的获得性耐药相关,但由于这些突变发生频率较小,获得的样本量较少,故其详细的作用机制还需进一步验证。
4.克服EGFR-TKI 耐药的策略
随着EGFR-TKI耐药分子机制的深入研究,多种抗EGFR-TKI耐药的药物逐渐进入临床试验阶段,并显示出显著的效果。不仅如此,多种治疗方式联合治疗,转换治疗等其他治疗方式也在进行越来越多的临床试验。总体而言,有以下几个方面可以帮助我们克服EGFR-TKI的耐药。
4.1 不可逆性TKI:多种不可逆性TKI如EKB-569、BIBW2992、CI-1033等正在被临床所应用。与传统的EGFR-TKI不同点在于不可逆转的EGFR-TKI共价结合于EGFR催化域ATP结合位点边缘的CYS-797,能明显抑制传统的EGFR-TKI的耐药。
4.2 多靶点抗肿瘤药物:NSCLC是高异质性的恶性肿瘤,单靶点药物在阻滞肿瘤驱动信号通路的同时也激活了肿瘤的逃逸机制,导致肿瘤细胞可能通过其他旁路再次激活增殖。因此,在确保低毒性、安全性的前提下可以尽可能多的抑制肿瘤信号通路。目前,多靶点的抗肿瘤药物,如舒尼替尼、索拉非尼、凡德他尼、阿西替尼等药物在NSCLC中的作用也正在进行各期临床试验。
4.3 针对c-Met 基因扩增的抑制剂:目前该抑制剂已进入临床试验阶段。
4.4 高效选择性EGFR突变体抑制剂: 一项I期临床研究发现,一种新的突变高效选择性EGFR-TKI药物AZD9291,对EGFR突变NSCLC患者或能提供一种新治疗选择。大约有50%患者出现肿瘤缩小,该药物对于T790M突变的患者(50%患者检测到)疗效尤其突出,而T790M突变是EGFR治疗耐药的主要原因,该药对于EGFR-TKI继发性耐药的患者无疑是一种福音。
5.结语
相对于传统化疗,EGFR-TKI靶向治疗更具优势,因其毒副作用弱,已成为晚期NSCLC的有效治疗手段之一。从基因学角度筛查,适合靶向治疗的患者能使治疗效果更佳。原发和获得性耐药等问题的出现使靶向治疗面临新的挑战,从而增加了临床治疗肺癌的难度。科学家正对此进行探索,已取得的成果正在帮助我们克服原发和获得性耐药的发生。未来的靶向治疗,可能会更加复杂,同时也会更加应用广泛。

公众微信二维码
关注"一生不得癌症秘诀"
微信/QQ:1151591580




